セディールは、住友製薬(現:大日本住友製薬)が1980年に創製し、1996年に販売開始した非ベンゾジアゼピン系の抗不安薬です。
Sedielは、ラテン語で「安静」を意味します。
抗不安作用は、もっぱらセロトニン1A受容体に対する作用に由来します。ベンゾジアゼピン受容体は関与しません。
そのため、ベンゾジアゼピン受容体を介した、筋弛緩作用、麻酔増強作用、自発運動抑制作用、協調運動抑制作用、抗けいれん作用をほとんど認めません。
また、眠気等の副作用も少なくなっています。
(ただし、添付文書上、自動車運転は控えることとされています)
眠気・めまい等が起こることがあるので、本剤投与中の患者には自動車の運転等危険を伴う機械の操作に従事させないように注意すること。(添付文書)
ベンゾ薬で問題となる依存性についても、精神依存性及び身体依存性ともに認められていません。
重大な副作用として、頻度は少ないですが肝機能障害、黄疸、セロトニン症候群、悪性症候群が報告されています。
効能・効果
・神経症における抑うつ、恐怖
・心身症(自律神経失調症、本態性高血圧症、消化性潰瘍)における身体症候ならびに抑うつ、不安、焦躁、睡眠障害
用法・用量
通常、1回10mgを1日3回服用します。最大60mg までの服用が可能です。
相互作用
①ベンゾ薬との併用で、相互に抗不安作用を増強します。
②ブチロフェノン系誘導体との併用で、抗ドパミン作用を軽度に増強します。
③カルシウム拮抗剤との併用で、降圧作用を軽度に増強します。
半減期等
単回投与
半減期:1.2時間(空腹)、1.4時間(食後)
最高血中濃度到達時間(Tmax):0.8時間(空腹)、1.4時間(食後)
最高血中濃度(Cmax):3.2ng/mL(空腹)、2.9ng/mL(食後)
反復投与
半減期:1.0±1.4時間
最高血中濃度到達時間(Tmax):0.8±0.6時間
最高血中濃度(Cmax):3.1±2.0 ng/mL
代謝
タンドスピロンは 主としてCYP3A4で代謝されます。また、CYP2D6 でもわずかに代謝されるようです。
副作用
使用成績調査等による副作用は、多い順に以下の通りです。
(合計6210例中)
嘔気・悪心・嘔吐・食欲不振 計59例(0.95%)
めまい・ふらつき(感) 計58例(0.93%)
倦怠(感)・脱力(感)・気分不良 計42例(0.68%)
ALT/AST/γ-GTP 上昇 計35例(0.56%)
頭痛・頭重(感) 計32例(0.52%)
もうろう状態 13例 (0.21%)
口渇 10例 ((0.16%)
以下、少し細かく作用機序等を見ていきます。
まず結合親和性ですが、セロトニン1A受容体に、選択的に作用します。
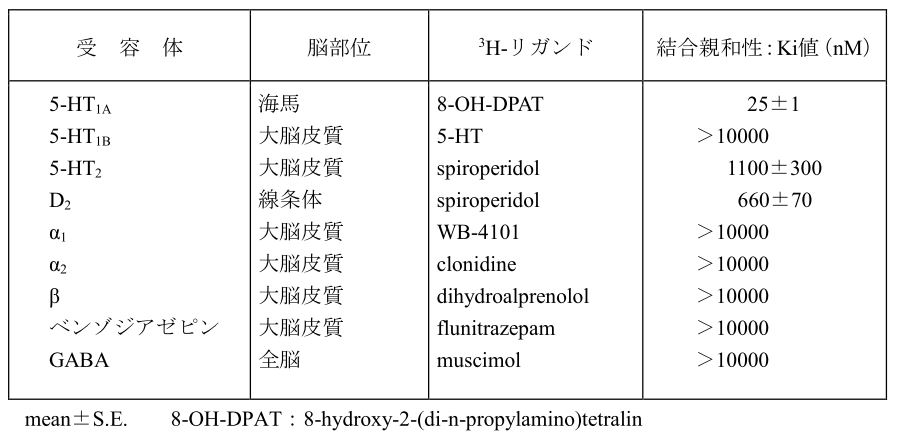
(IFより)
セロトニン1A受容体には、シナプス前受容体とシナプス後受容体がありますが、タンドスピロンの作用の多くはシナプス後1A受容体に対するパーシャルアゴニスト作用で説明されています。
セロトニン1A受容体
シナプス前受容体:自己受容体で、セロトニン放出を抑制します。セディールはフルアゴニストとして作用します。
シナプス後受容体:セディールはパーシャルアゴニストとして作用し、1A受容体を活性化します。
シナプス後のセロトニン1A受容体の活性化は、過分極(抑制)効果をもたらします(Andrad)。シナプス後の2A受容体の活性化が脱分極(興奮)効果をもたらすのと対照的です。
また、通常の状態の脳では、大脳皮質および辺縁系領域のセロトニン機能を支配しているのは1A受容体であると考えられています(Puig)。
極端なストレス下では、1A受容体はダウンレギュレートされ(Berton)、2A受容体はアップレギュレーションされると考えられています(Benekareddy)。
脳内のセロトニン伝達が減少すると、衝動的で攻撃的な行動が増加します(Audero)。セロトニンによる攻撃性と衝動性の緩和効果は、シナプス後1A受容体のシグナルによって媒介されていると考えられています(Sanchez and Hyttel)。
シナプス後1A受容体は、辺縁系領域、特に海馬に高密度に発現しています(Pazos and Palacios)。1A受容体を刺激することで、錐体細胞の活動は抑制されます。つまり、シナプス後1A受容体の抑制作用が、大脳辺縁系の過活動を抑制することで不安が軽減されると考えられています(Dongら、1998)。
タンドスピロンやSSRIの持続投与は、辺縁系の反応性の低下をもたらすことで、不安を軽減します。
衝動性の緩和に関連して、タンドスピロンの投与が摂食障害に有効であったという報告があります(Okita)。
パニック障害患者はセロトニン1A受容体の機能が低下していることが示されています[Lesch]。 さらに、未治療のパニック障害患者において5-HT1A結合が減少していることが明らかになっています[Nash,Neumeister]。
タンドスピロンは、PTSDの動物モデルで恐怖の消去を促進することで不安を軽減するという治療効果を示しました。
ただし、不安の治療におけるタンドスピロンの主な欠点は、短い半減期と1週間または2週間の遅発性です[Godbout]。これは、シナプス前1A受容体の脱感作に時間がかかるためです。
最近行われた無作為化非盲検試験において、若年者の社会不安障害の治療においてタンドスピロンがセルトラリンと比較して非劣性であることが示されています[Huang]。
以上、タンドスピロンは、PTSDやSADの治療において、有効な治療薬となる可能性があります。
うつ病では、MAOIや三環系抗うつ薬、SSRI、リチウム、バルプロ酸などは、直接・間接にシナプス後1A受容体シグナルを増加させます[Savitz]。
また死後研究で、自殺患者では1A受容体の数が減少していることが判明しており、PETスキャン分析により1A受容体の結合親和性の低下が示されています[Savitz]。
さらに、1A受容体ノックアウトマウスを用いた研究により、1A受容体の機能障害がうつ病の根本的なメカニズムであるという仮説が立てられています[Savitz]。
うつ病の緩和には、セロトニン受容体の中でも特に1A自己受容体の脱感作が重要で、これを介してセロトニンが役割を果たすという説が唱えられています[Schatzberg,Drago]。
抗精神病薬による大脳皮質でのドパミン放出には、1A受容体の活性化が必要であることが、ノックアウトマウスを用いた研究で確認されました[Newman-Tancredi]。
1A受容体のパーシャルアゴニストであるタンドスピロンは、ハロペリドールで治療を受けた統合失調症患者の陰性症状を軽減します 。
その効果は、ドーパミン神経ループを介して間接的に皮質のドーパミン神経伝達を増強することによると考えられています[Saito、Li]。
抗精神病薬のうち、クロザピン、クエチアピン、(ジプラシドン–本邦未発売)、アリピプラゾール、ブレクスピプラゾールは1Aパーシャルアゴニストです。
シナプス後1A受容体は、タンドスピロンを繰り返し投与しても脱感作されません[Blier]。
縫線核におけるシナプス前5-HT1A自己受容体の脱感作の持続時間が、タンドスピロン治療の作用の開始が遅いことを説明します。
いくつかの臨床試験において、タンドスピロンとSSRIを併用することで、1A自己受容体の脱感作が相乗的に促進され、その結果、より迅速な作用発現や抗うつ作用の増強が認められています。
ある症例報告では、タンドスピロンの投与で、前頭前野のドーパミン放出が増加したことが示されています[Kishi]。
1A受容体は錐体外路性運動症状などの運動機能の調節に関与していることも示されています。タンドスピロンは前臨床試験および臨床試験で抗パーキンソン病作用を発揮し、約40%のパーキンソン病患者において、歩行の安定性と日常生活の活動を改善しました[Nomoto]。
このタンドスピロンの改善効果は、1A受容体拮抗薬でほぼ完全に拮抗されましたが、D2拮抗薬であるハロペリドールでは拮抗されなかったので、タンドスピロンの抗パーキンソン作用は、ドーパミン作動性とは無関係に1A受容体を直接活性化することに関連していると考えられます。
1A受容体アゴニストは、皮質-線条体グルタミン酸経路を阻害し、線条体における細胞外グルタミン酸レベルを低下させます。タンドスピロンはシナプス後受容体を活性化した後、皮質-線条体のグルタミン酸経路を阻害してグルタミン酸の放出を減少させることで、運動機能障害を改善すると考えられます。
またタンドスピロンは用量依存的に、統合失調症患者の衝動的な行動を抑制することが示されています[Ohmura]。